

SIDB PhD student Nawon Kim (Cousin lab) and colleagues, have recently published a paper, looking into the effects of inactivating or ‘knocking out’ the Fmr1 gene, on nerve ending recycling. It is this same gene that is mutated (and doesn’t function properly) in individuals with fragile X syndrome.
What did they find out?
Nawon works on dysfunctional synaptic vesicle (SV) recycling which is essential in maintaining the function of presynapses (the end of a synapse which sends a signal to another synapse to ‘communicate’). In this new paper, the team discovered that a mode of endocytosis – a mechanism for replenishing the SVs in the nerve terminals – was activated later in neurons (brain cells) where the Fmr1 gene was knocked out, compared to typical (or ‘wild type’) neurons. This mode of endocytosis is called activity-dependent bulk endocytosis (ADBE), and is a pathway which sustains brain communication during high activity.
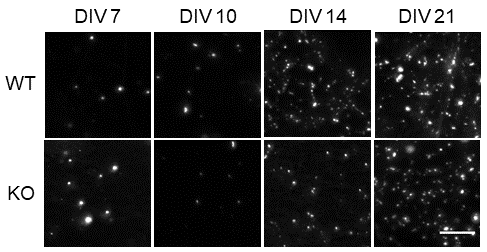
Why is this important?
Their findings are important as they imply that the delayed recruitment of ADBE is a potential contributing factor in the development of circuit dysfunction in fragile X syndrome, and potentially other neurodevelopmental disorders.
What’s next?
Nawon and colleagues will now aim to find out the molecular mechanism behind the developmental delay of the Fmr1 knockout neurons, and try to understand why these neurons are delayed and how they catch up later. They will also look at whether this delayed development is a common feature in other models of autism.
Source:
Read the full paper in Journal of Neurochemistry:
Delayed recruitment of activity-dependent bulk endocytosis in Fmr1 knockout neurons
Nawon Kim, Katherine Bonnycastle, Peter C. Kind, Michael A. Cousin
DOI: 10.1111/jnc.16178
Earlier work
The Cousin lab published an earlier paper that revealed a specific defect in ADBE in neurons that model fragile X syndrome. Molecular studies revealed that the reason for this dysfunction was due to faulty binding of fragile X messenger ribonucleoprotein 1 to channels that usually correct cell hyperexcitability (BK channels). Activation of these channels corrected the ADBE deficit, suggesting this may be a valuable therapeutic avenue for further investigation.
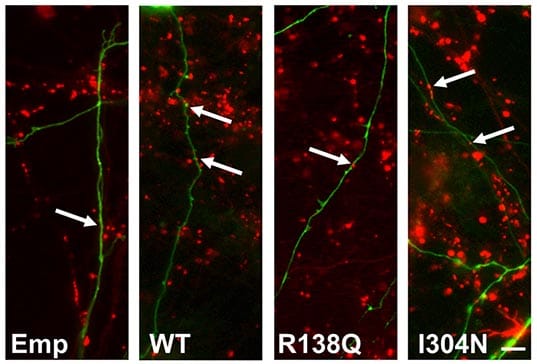
Source:
Read the full paper in Journal of Neuroscience:
FMRP Sustains Presynaptic Function via Control of Activity-Dependent Bulk Endocytosis.
Katherine Bonnycastle, Peter C. Kind, Michael A. Cousin
DOI: 10.1523/JNEUROSCI.0852-21.2021.
